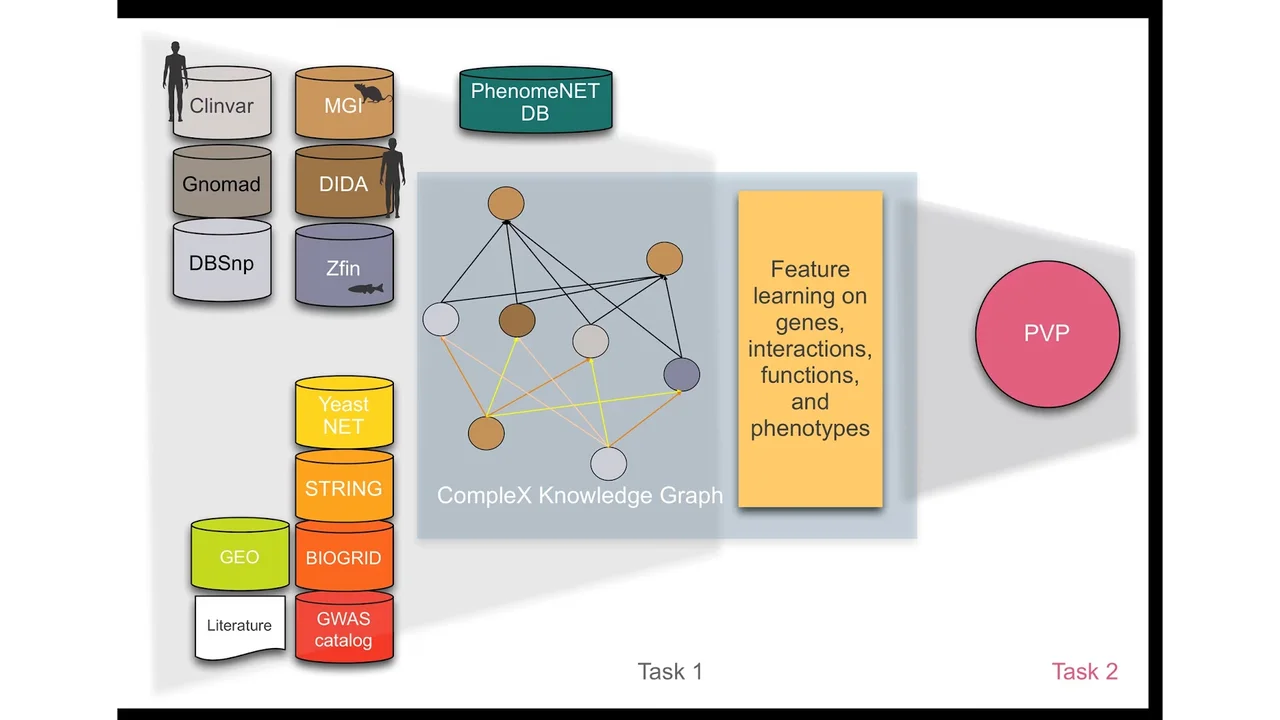
Phenotypes Research OverviewCompleX: Variant prioritization in complex diseaseDetailsWe are now at a stage when the discovery of new disease genes is slowing. The number of patients remaining undiagnosed following whole exome sequencing argues either that many more disease genes and variants still await discovery, or that the heterogeneity and novelty of disease phenotypes that we see are due to a combination of alleles of multiple, known, disease genes in the same individual. While the particular combinations of alleles in the same person may be rare, they likely involve medium-rare or common alleles as well as rare ones.With increasing evidence that many rare diseases have oligo- or polygenic origins, it is important to develop new methods that can be applied to individuals to provide molecular diagnosis in a personalized manner; traditional Mendelian genetic approaches ignore the contribution of medium-rare to common alleles, and population-level approaches generally have insufficient power to identify the variants contributing to polygenic diseases which involve both low frequency and common variants. We believe that our phenotype-driven, knowledge-based approach provides a technology for the breaking of this impasse which is necessary for the realization of the power of personalized medicine. The major impact of our work will be on developing diagnostic support tools for common and genetically complex diseases which are currently the greatest contributors to morbidity and mortality outside the tropical regions. In many countries, we see an emerging pattern of rapidly increasing multichronic diseases and co-morbidities, linked through genetic and environmental risk factors. These have profound consequences for economic productivity and healthcare costs. Consequently, it is important to be able to understand why specific individuals develop or fail to develop particular co-morbidities or different spectra of disease manifestation, how this impacts treatment decisions, and what might be the genetic risk factors for disease outcomes.Phenotype-based methods have repeatedly shown to be highly effective in identifying causative variants in whole genome or whole exome sequences. The main limitation of phenotype-based methods, however, is the limited availability of characterized genotype-phenotype associations. Model organism phenotypes have in the past been used to supplement genotype-phenotype associations observed in humans and were demonstrated to predict disease genes. Nevertheless, in almost all cases, genotypes are loss-of-function or gain-of-function variants in single genes. Consequently, phenotypes that arise specifically from abnormal functioning of two or more genes in the same individual are not commonly captured; in the cases in which complex genotypes and their associations with phenotypes are recorded (e.g., in the mouse and fish model organism databases), they are not integrated, not distinguished by the type of interaction between variants, and cannot systematically be queried.CollaboratorsPaul Schofield, University of CambridgeGeorge Gkoutos, University of Birmingham