Sequencing and computational analysis of MRSA samples
Overview
Methicillin-resistant Staphylococcus aureus (MRSA) is a leading cause of hospital-acquired infection worldwide, and Saudi Arabia presents a singular epidemiological setting: the Kingdom hosts more than two million Hajj pilgrims and over twenty million Umrah pilgrims annually, drawn from every region of the Muslim world. Mass gatherings of this scale offer a natural experiment in how human mobility shapes bacterial population structure and the dissemination of antimicrobial resistance, but until recently nationally representative genomic data have been lacking. The project was carried out as a joint KAUST-KACST program led by Hoehndorf and Mohammed Al-Fageeh (KACST) with co-investigators Takashi Gojobori and Vladimir Bajic, and brought together more than thirty hospitals and reference laboratories across the country (KACST-KAUST award ETSC-2018-05-27-01).
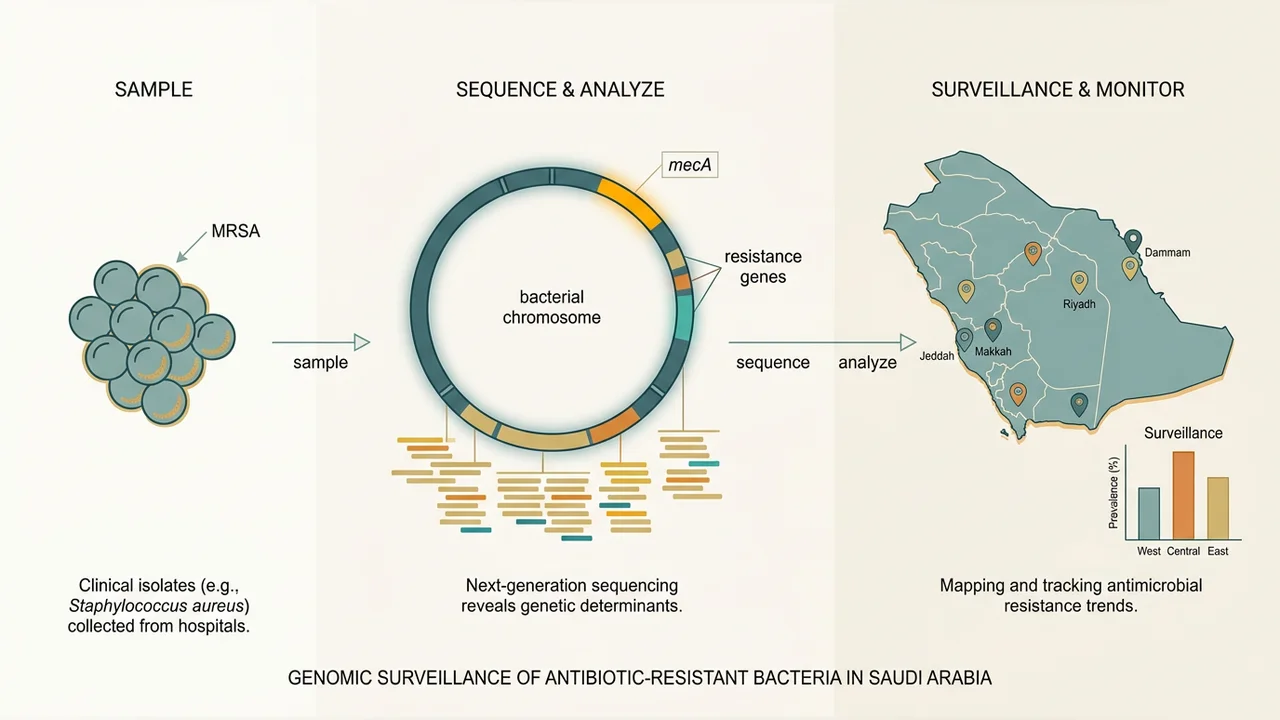
The completed work delivered the first nationwide, integrated genotype-phenotype survey of S. aureus in Saudi Arabia. The team assembled a collection of 686 S. aureus isolates from seven regions (Riyadh, Jeddah, Makkah, Madinah, Hail, AlHasa, and Jazan), comprising clinical samples from tertiary care hospitals, community-screening isolates from healthy individuals, and wastewater samples. Each isolate was whole-genome sequenced on Illumina NovaSeq, phenotyped on a 16-antibiotic VITEK 2 panel, and described using a FAIR-compliant RDF metadata model built on standard ontologies (NCBI Taxonomy, ChEBI, GENEPIO, NCIT, UO). The complete analysis was implemented as a Common Workflow Language pipeline and executed on Arvados, with all sequences, phenotypes, and workflow definitions deposited in ENA (PRJEB59751) and Zenodo so that the dataset is fully reproducible and reusable.
The resulting manuscript — Genomic diversity and antimicrobial resistance of Staphylococcus aureus in Saudi Arabia: a nationwide study using whole-genome sequencing — reports several findings that change the picture of MRSA in the region. Pilgrimage cities (Jeddah, Makkah, Madinah) carry significantly greater genetic diversity, more clonal complexes, and higher overall resistance rates than non-pilgrimage regions, and harbour internationally recognized clones (the Bengal Bay CC1-ST772, the South Pacific CC30-ST30, ACME-positive USA300-like lineages) consistent with importation along Hajj and Umrah routes. Fourteen previously undescribed sequence types were identified, the majority entering through Jeddah, the principal international entry point. Genomic prediction of resistance from CARD, ResFinder, and DeepARG reached high precision and recall for beta-lactams and fusidic acid but performed poorly for aminoglycosides and macrolides, demonstrating concretely that genotype-only surveillance is currently insufficient for clinical decision-making and that phenotypic testing remains necessary alongside sequencing. A pangenome-wide association analysis recovered canonical determinants (mecA, mecR1, ermC, msrA, tet(K)) and surfaced non-canonical associations whose interpretation will require further work.
Beyond the publication, the project leaves a reusable infrastructure for genomic surveillance in mass-gathering settings: a curated reference collection, an RDF data model that makes S. aureus metadata interoperable with other surveillance datasets, and a reproducible pipeline that can be redeployed on new isolates. The findings argue concretely for wastewater-based surveillance at pilgrimage airports and ablution facilities as an early-warning mechanism for emerging resistant lineages, and provide the baseline against which any such system would be measured.
Period: 2018–2021
Funding
- King Abdulaziz City for Science and Technology
— Grant ID:
RGC/3/3689-01-01(PI) — USD 362,159
Team
- Robert Hoehndorf — PI (KAUST (Professor of Computer Science))
- Mohammed Al Fageeh — PI (KACST)
- Takashi Gojobori — CoI (KAUST (formerly CBRC, now dissolved))
- Vladimir Bajic — CoI (Former KAUST CBRC director (retired; CBRC now dissolved))
Software
- PhenomeNET-VP — Phenotype-driven variant prioritization for whole-exome and whole-genome sequencing data; widely used implementation of the phenotype-aware variant ranking approach. https://github.com/bio-ontology-research-group/phenomenet-vp
Topics: Genomics, Microbial communities
